Search results
Search for "quantum chemical calculations" in Full Text gives 87 result(s) in Beilstein Journal of Organic Chemistry.
Beyond n-dopants for organic semiconductors: use of bibenzo[d]imidazoles in UV-promoted dehalogenation reactions of organic halides
Beilstein J. Org. Chem. 2023, 19, 1912–1922, doi:10.3762/bjoc.19.142

- potentials of the halides that can be reduced in this way, quantum-chemical calculations, and steady-state and transient absorption spectroscopy suggest that UV irradiation accelerates the reactions via cleavage of the dimers to the corresponding radical monomers. Keywords: dehalogenation; n-dopant







A deep-red fluorophore based on naphthothiadiazole as emitter with hybridized local and charge transfer and ambipolar transporting properties for electroluminescent devices
Beilstein J. Org. Chem. 2023, 19, 1664–1676, doi:10.3762/bjoc.19.122

- points were measured using a Krüss KSP1N melting point meter and are uncorrected. Quantum chemical calculations were executed using the Gaussian 16 package [67]. Density functional theory (DFT) calculations at the B3LYP level of theory with the 6-31G(d,p) basis set were performed to realize the ground









Organophosphorus chemistry: from model to application
Beilstein J. Org. Chem. 2023, 19, 89–90, doi:10.3762/bjoc.19.8

- chemistry and beyond. Quantum chemical calculations are a great support for organic chemists when exploring structures, reactivities, and mechanisms. In this thematic issue, the Diels–Alder cycloaddition of 2-phosphaindolizine, 1-aza-2-phosphaindolizine, 3-aza-2-phosphaindolizine, and 1,3-diaza-2
Redox-active molecules as organocatalysts for selective oxidative transformations – an unperceived organocatalysis field
Beilstein J. Org. Chem. 2022, 18, 1672–1695, doi:10.3762/bjoc.18.179

- coupling of alkylarenes with NHPI was realized recently [94] (Scheme 11). In this process the formation of benzylic radicals is catalyzed by PINO radicals. According to the mechanism supported by quantum chemical calculations, the mixture of C–O and C–N products is formed as a result of the attack of the








































Naphthalimide-phenothiazine dyads: effect of conformational flexibility and matching of the energy of the charge-transfer state and the localized triplet excited state on the thermally activated delayed fluorescence
Beilstein J. Org. Chem. 2022, 18, 1435–1453, doi:10.3762/bjoc.18.149

- state lifetimes are 45 μs and 108 μs in HEX and ACN, respectively (Figure S36, Supporting Information File 1). For NI-PTZ-O, 3LE and 3CT states were observed in ACN, and the lifetime was determined as 71 μs (Figure S35c). Computational investigations To explain the experimental results, quantum chemical
- calculations were used to obtain additional insights into both the excited states involved and the photo-deactivation dynamics. First, the ground state geometry of the compounds was optimized (Figure 9). For the compact NI-PTZ and NI-PTZ-O dyads, the two units adopt almost orthogonal geometry. A similar result
Characterization of a new fusicoccane-type diterpene synthase and an associated P450 enzyme
Beilstein J. Org. Chem. 2022, 18, 1396–1402, doi:10.3762/bjoc.18.144

- . Subsequently, the extensive NMR analysis established the planar structure of 1, and its relative configuration was partially assigned as 2S*,3S*,6R*,10R* by the NOESY spectrum (Supporting Information File 1, Table S3 and Figures S3–S8). As to the stereochemistry of C11, quantum chemical calculations of 13C NMR
- ). For isolation of 3, the mutated tadA, along with GGPPS gene, was introduced into A. oryzae NSAR1, and the resulted transformant produced 3 at a titer of 0.6 mg/L (Figure 3A, line iv). By comparison of NMR data and specific optical rotation values, together with quantum chemical calculations of 13C NMR





Ferrocenoyl-adenines: substituent effects on regioselective acylation
Beilstein J. Org. Chem. 2022, 18, 1270–1277, doi:10.3762/bjoc.18.133

- approach of an electrophile (e.g., FcCOCl) towards the N7 atom. In the course of N9-isomer formation no similar steric hindrance is encountered. This is supported by our quantum-chemical calculations which compared the two transition state structures for the ferrocenoylation of the N6,N6-di-tert







Derivatives of benzo-1,4-thiazine-3-carboxylic acid and the corresponding amino acid conjugates
Beilstein J. Org. Chem. 2022, 18, 1195–1202, doi:10.3762/bjoc.18.124

- better results, but the oxidative dimerization was still a competitive reaction. NMR analyses of the isolated product 10ac also revealed the presence of the corresponding 2H-isomer in a low amount, but it was not isolated separately. Quantum chemical calculations (ωB97xD/6-31G(d)//MN15/6-311+G(2d,p
Understanding the competing pathways leading to hydropyrene and isoelisabethatriene
Beilstein J. Org. Chem. 2022, 18, 972–978, doi:10.3762/bjoc.18.97

- two byproducts, isoelisabethatrienes A and B. Fascinatingly, a single active site mutation (M75L) diverts the product distribution towards isoelisabethatrienes A and B. In the current work, we study the competing pathways leading to these products using quantum chemical calculations in the gas phase





Mechanochemical halogenation of unsymmetrically substituted azobenzenes
Beilstein J. Org. Chem. 2022, 18, 680–687, doi:10.3762/bjoc.18.69

- species, cyclopalladated intermediates, and products (Figure 1). The monitoring results confirmed the crucial role of TsOH and acetonitrile (MeCN) as additives in the catalytic bromination of the C–H bond in L1. The experimental results were supported by quantum-chemical calculations, which showed that








Substituent effect on TADF properties of 2-modified 4,6-bis(3,6-di-tert-butyl-9-carbazolyl)-5-methylpyrimidines
Beilstein J. Org. Chem. 2022, 18, 497–507, doi:10.3762/bjoc.18.52

- confirm the structures of the synthesized compounds (see Figures S1–S27 in Supporting Information File 1). DFT analysis To assess the structural and electronic properties of the chromophores tCbz-mPYR and 1–6, quantum chemical calculations were performed. DFT analysis revealed that all studied compounds
- 49 ms integration time. Solid-state samples were mounted in a closed cycle He cryostat (Cryo Industries 204 N) for PL measurements in oxygen-free conditions. Quantum chemical calculations were carried out by using density functional theory at the B3LYP/6-31G(d) level as implemented in a software






2,4-Bis(arylethynyl)-9-chloro-5,6,7,8-tetrahydroacridines: synthesis and photophysical properties
Beilstein J. Org. Chem. 2021, 17, 1629–1640, doi:10.3762/bjoc.17.115

- . In order to elucidate these experimental observations, quantum chemical calculations based on density functional theory (DFT) methodology were performed. The estimated visualization of highest occupied and lowest unoccupied molecular orbitals, as well as the molecular electrostatic potential (MEP) of






Recent advances in the application of isoindigo derivatives in materials chemistry
Beilstein J. Org. Chem. 2021, 17, 1533–1564, doi:10.3762/bjoc.17.111

- the efficiency from 4.58 to 6.21%. Based on the data of quantum chemical calculations, Park et al. [34] showed that in the fluorinated derivative, the dihedral angle between the thiophene rings is 0.88° (for comparison, in compound 22a it is 17.55°), which provides better planarity of the polymer and

































Mesoionic tetrazolium-5-aminides: Synthesis, molecular and crystal structures, UV–vis spectra, and DFT calculations
Beilstein J. Org. Chem. 2021, 17, 385–395, doi:10.3762/bjoc.17.34

- -butyltetrazolium-5-aminide, its N,N’-ethylene-bridged bis-derivative and (1,3-di-tert-butyl-1H-tetrazol-3-ium-5-yl)(1-phenyl-1H-tetrazol-5-yl)amide by single crystal X-ray analysis. The structural and spectral features of the tetrazolium-5-aminides are discussed by using quantum-chemical calculations. Keywords
- -butyltetrazolium-5-aminide, its N,N’-ethylene-bridged bis-derivative and (1,3-di-tert-butyl-1H-tetrazol-3-ium-5-yl)(1-phenyl-1H-tetrazol-5-yl)amide by single crystal X-ray analysis. The structural and spectral features of the tetrazolium-5-aminides were discussed by using quantum-chemical calculations











Muyocopronones A and B: azaphilones from the endophytic fungus Muyocopron laterale
Beilstein J. Org. Chem. 2020, 16, 2100–2107, doi:10.3762/bjoc.16.177

- experimental ECD spectra to elucidate the absolute configuration. In the quantum-chemical calculations, the generation of an excessive number of conformers was avoided using a molecular model in which the β-hydroxycarboxylic acid side chain at the C-7 position was simplified to an acetyloxy group (Figure 4A






One-pot synthesis of oxazolidinones and five-membered cyclic carbonates from epoxides and chlorosulfonyl isocyanate: theoretical evidence for an asynchronous concerted pathway
Beilstein J. Org. Chem. 2020, 16, 1805–1819, doi:10.3762/bjoc.16.148
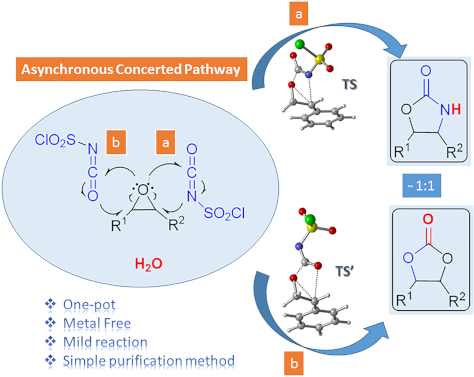
- computational studies, such reactions of isocyanates may proceed through a concerted pathway. The remaining uncertainties in the mechanisms of the similar reactions inspired us to carry out quantum chemical calculations for the formation of oxazolidinone and five-membered cyclic carbonates. Results and















Rearrangement of o-(pivaloylaminomethyl)benzaldehydes: an experimental and computational study
Beilstein J. Org. Chem. 2020, 16, 1636–1648, doi:10.3762/bjoc.16.136

- ), respectively, was also investigated by DFT level quantum chemical calculations with the same parameters as in the case of the 1→2 rearrangement. The racemic RS and RR forms were taken into consideration, and the computations showed that the formation of the RS compound required an energy investment of ΔG#4e













Oxime radicals: generation, properties and application in organic synthesis
Beilstein J. Org. Chem. 2020, 16, 1234–1276, doi:10.3762/bjoc.16.107

- example, based on N-containing heterocycles (isoxazolones, pyrazolones, pyrazolidin-3,5-diones, and 1,2,3-triazolones [52]), sulfones [65], and phosphonates [54] (Scheme 8). Based on the data of EPR spectroscopy [35][38][49][50][66] and quantum chemical calculations [67], the maximum spin density in
- isomers (E and Z) exist. The isomerization of oxime radicals proceeds much easier than for the corresponding oximes; the observation of individual isomers is generally possible only at low temperatures [68][69] (about 190 K). According to quantum chemical calculations, the oxime radicals have an increased









































































The charge-assisted hydrogen-bonded organic framework (CAHOF) self-assembled from the conjugated acid of tetrakis(4-aminophenyl)methane and 2,6-naphthalenedisulfonate as a new class of recyclable Brønsted acid catalysts
Beilstein J. Org. Chem. 2020, 16, 1124–1134, doi:10.3762/bjoc.16.99

- providing supercomputer facilities. Funding The X-ray diffraction data were collected with financial support from the Ministry of Science and Higher Education of the Russian Federation using the equipment of the Center for Molecular Composition Studies of INEOS RAS. Quantum chemical calculations were










Preparation of 2-phospholene oxides by the isomerization of 3-phospholene oxides
Beilstein J. Org. Chem. 2020, 16, 818–832, doi:10.3762/bjoc.16.75

- double bond migration pathways were elucidated by quantum chemical calculations. Keywords: chlorophosphonium salts; isomerization; 2-phospholene oxides; 3-phospholene oxides; quantum chemistry; Introduction P-Heterocyclic derivatives are valuable targets in synthetic organophosphorus chemistry [1][2][3
- chemical calculations at MP2/6-31++G(d,p) and MP2/6-311++G(2d,2p) levels of theory, including the PCM solvent model with the parameters of THF. For these investigations, the 1-phenyl or 1-ethyl-3-methylphospholene oxides (1a or 1h) were considered as model compounds, but phospholene oxides incorporating
- isomerization remained incomplete under these conditions. It was found that methyl group(s) at positon 3 or 4 in the P-heterocyclic ring hinder the isomerization in all reaction pathways investigated. The mechanisms of acid- or base-mediated, as well as thermal isomerizations were elucidated by quantum chemical







Photophysics and photochemistry of NIR absorbers derived from cyanines: key to new technologies based on chemistry 4.0
Beilstein J. Org. Chem. 2020, 16, 415–444, doi:10.3762/bjoc.16.40

- withdrawing moieties. Quantum chemical calculations showed only small contributions of the substituent placed at the meso-position to the electron density in the HOMO/LUMO pattern [81]. Presumably, coupling of lower occupied molecular orbitals with the HOMO on the one hand side and higher unoccupied MOs with























The reaction of arylmethyl isocyanides and arylmethylamines with xanthate esters: a facile and unexpected synthesis of carbamothioates
Beilstein J. Org. Chem. 2020, 16, 159–167, doi:10.3762/bjoc.16.18

- reactions, a mechanism is proposed in which the key steps are supported by quantum chemical calculations. Keywords: benzylamines; carbamothioates; density functional theory; intrinsic reaction coordinate analysis; isocyanides; sodium hydride; xanthate esters; Introduction Carbamothioates (thiocarbamates
- hypothesis. Computational studies on the proposed reaction mechanism Several possible reaction mechanisms were considered to account for the unexpected products obtained. Ultimately, we employed quantum chemical calculations to shed light on the most probable reaction pathway for the observed products, as
- simplify the quantum chemical calculations, the reactions shown in Scheme 2 involve a hydride as the nucleophile or base [34], although it is possible that dimethylamide, formed from the reaction of sodium hydride with DMF [35], could be the initiating nucleophile/base. All computations were carried out








Why do thioureas and squaramides slow down the Ireland–Claisen rearrangement?
Beilstein J. Org. Chem. 2019, 15, 2948–2957, doi:10.3762/bjoc.15.290

- quantum-chemical calculations employing long-range corrected hybrid density ωB97X-D functional [42]. This dispersion-corrected functional displays very balanced overall performances and has demonstrated excellent treatment of noncovalent interactions [43], which are very important in our studied system









Azologization and repurposing of a hetero-stilbene-based kinase inhibitor: towards the design of photoswitchable sirtuin inhibitors
Beilstein J. Org. Chem. 2019, 15, 2170–2183, doi:10.3762/bjoc.15.214

- ortho methyl groups in 2f, intramolecular photocyclization could be prevented. To verify the hypothetical structures derived from irradiation of 2b, we carried out quantum chemical calculations of the double bond isomers (E)-2b and (Z)-2b as well as the oxidized compounds 8a and 8b. We used density










Tautomerism as primary signaling mechanism in metal sensing: the case of amide group
Beilstein J. Org. Chem. 2019, 15, 1898–1906, doi:10.3762/bjoc.15.185

- 5. The quantum-chemical calculations for 4 and 5 have demonstrated that the stable enol tautomers exist as intramolecular C=O···HO bonded system, while in the K forms the ionophore part does not participate in hydrogen bonding and can be considered as a basic 2-alkyl substitution [9]. Consequently
- still not appear even after 1024 scans. Theoretical calculations Quantum-chemical calculations were performed using the Gaussian 09 D.01 program suite [17]. The M06-2X functional [18][19] was used with the 6-31++G** basis set for the calculations. This fitted hybrid meta-GGA functional with 54% HF








